Patient Protection in Clinical Trials

How are my rights and privacy protected in a clinical trial?
All modern research that involves people – including clinical trials – must be conducted according to the values of Respect, Beneficence, and Justice. Several safeguards protect the rights, safety and privacy of volunteers who participate in clinical trials as much as possible. International and federal guidelines are in place to ensure clinical trials are conducted ethically, and specialist committees of experts scrutinize individual trial details before and during a study. Through a formal process called informed consent, anyone considering joining a clinical trial must be fully informed by the research team about the study details, including its purpose, all known risks and potential benefits, how privacy will be protected, and who will have access to participant information.
What does it mean to be randomized?
In randomized clinical trials, a computer randomly divides patients into separate groups that are given different treatments or interventions (for example, one group is given a new experimental treatment and another group an approved standard treatment). The patients are separated by chance, which means that if you participate in a randomized study, you and your doctor will not be able to choose which treatment you receive. Dividing participants into groups randomly means that the groups will be similar and the different treatments or interventions can be compared more fairly.
Some randomized trials include a control group that does not receive the new treatment being studied. This group is compared to a group of people who receive the new treatment to see how well the new treatment works and if there are differences in side effects. Although people in the control group do not get the new treatment, they typically receive the best available treatment that is currently approved for their condition. When deciding whether to participate in a randomized trial, you should be comfortable with the possible side effects and expected benefits of all possible treatments in the study, since you and your doctor will not be able to choose which group you are assigned to.

Placebos in Clinical Trials
A placebo is an inactive substance that looks like medicine but is not. Nowadays, most randomized clinical trials in melanoma involve giving active medicine (not placebos) to all groups of patients, whether it is a new treatment or the best available treatment that has already been approved (known as the “standard of care”). Placebo may be given alone only if there is no available standard treatment, but it is considered ethically unacceptable to give people placebo alone in a clinical trial when there is an effective treatment for their condition. Because there are several available standard treatments for melanoma, placebos are not typically used alone in melanoma trials.
If a placebo is given to patients in a melanoma trial, it is usually given together with the best available treatment. In trials evaluating a combination of two or more treatments, for example, patients in the experimental arm may receive a standard treatment together with a new experimental treatment, versus participants in the control group that may be given a placebo together with the standard therapy. They would not be given placebo only.
Placebos in Clinical Trials
A placebo is an inactive substance that looks like medicine but is not. Nowadays, most randomized clinical trials in melanoma involve giving active medicine (not placebos) to all groups of patients, whether it is a new treatment or the best available treatment that has already been approved (known as the “standard of care”). Placebo may be given alone only if there is no available standard treatment, but it is considered ethically unacceptable to give people placebo alone in a clinical trial when there is an effective treatment for their condition. Because there are several available standard treatments for melanoma, placebos are not typically used alone in melanoma trials.
If a placebo is given to patients in a melanoma trial, it is usually given together with the best available treatment. In trials evaluating a combination of two or more treatments, for example, patients in the experimental arm may receive a standard treatment together with a new experimental treatment, versus participants in the control group that may be given a placebo together with the standard therapy. They would not be given placebo only.
Sometimes, randomized trials are “blinded,” meaning that the patients, or the patients along with their doctors, do not know which treatment they are being given. This can help reduce bias, which can happen when patients or doctors have preconceptions about how safe or effective a drug is (whether conscious or unconscious). If there is an emergency, doctors can request to know what treatment their patient is receiving.
Who makes sure patients’ rights are protected?
Researchers must follow regulatory requirements for clinical trials that protect patient rights. A number of international and federal guidelines are in place to make sure trials are conducted ethically. For example, an international standard known as Good Clinical Practice (GCP) sets ethical and scientific quality standards for designing and conducting clinical trials. In the U.S., government organizations including the Department of Health and Human Services, Food and Drug Administration (FDA), and National Institutes of Health (NIH) are involved in setting rules, providing advice, and monitoring different types of clinical trials. For example, the FDA sets rules for clinical trials that involve experimental drugs or devices.
Specialist groups provide oversight for individual clinical trials to make sure the volunteers who participate are protected:
Institutional review boards (IRBs) are independent committees of doctors, statisticians and community members who make sure a given trial is ethical and that participant rights are being protected. In the U.S., IRBs approve and supervise all studies involving testing of a drug, biological product, or medical device in people. IRBs review the clinical trial protocol, or detailed plan for the study, to confirm that it addresses a worthwhile question and ensures the safety of participants. IRBs also review the informed consent document to make sure it is accurate, complete, and easy to understand. While IRBs are mainly involved in approving the study before it begins, they continue to monitor the study to ensure it is performed as agreed to in the protocol. You can contact the IRB with questions or concerns if you are in a clinical study.
Data Safety Monitoring Boards (DSMBs) watch the progress and results of a trial while it is happening. A DSMB can decide to stop a trial early if they can see that new treatment is much more (or much less) effective than the control arm/current standard of care. If your trial is stopped for this reason, you would get the better treatment no matter what you were originally assigned to. A DSMB can also stop a clinical trial early due to safety concerns, particularly if it becomes clear that the risks are much greater than the benefits of treatment. This prevents people from being exposed to more possible harm.
What is informed consent?
Informed consent as a legal, regulatory, and ethical concept, is an integral part of research. In clinical trials, informed consent is the process of providing all relevant information about the trial’s purpose, risks, benefits, alternatives, and procedures to you as a potential participant. You can then make an informed decision about whether or not to participate, considering your own personal interests and circumstances.
The informed consent process provides people with ongoing explanations that will help them make educated decisions about whether to continue participation in a clinical trial. The process does not end with the signing of informed consent documents. If new benefits, risks, or side effects are discovered during the trial, researchers must inform participants. Participants are encouraged to ask questions at any time.
Before agreeing to take part in a clinical trial, participants have the right to learn everything that is involved in the trial – including all details about treatment, tests, and possible risks and benefits. The information has to be in a format and language that can be easily understood.
What to look for during informed consent:
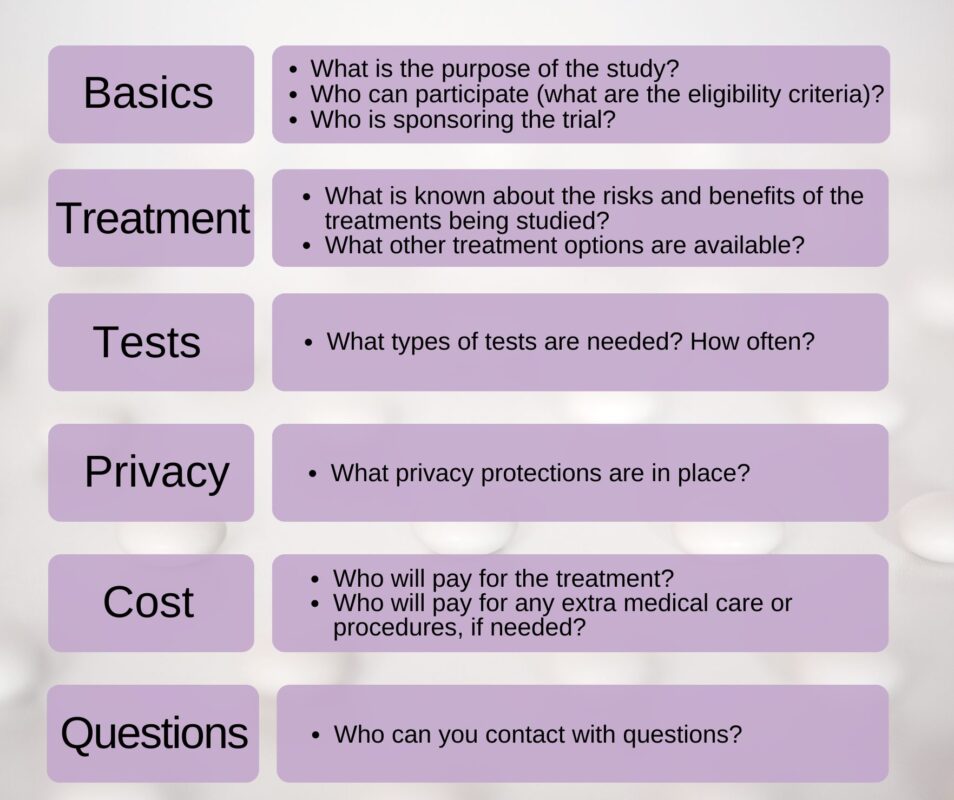
How will my health information be kept private?
Your personal and health information will be kept confidential as much as possible during a clinical trial. As would happen during normal care outside of a clinical trial, your healthcare team will have access to your personal and medical information so they can treat you.
If you agree to be in a clinical trial, you may be asked to let your healthcare team share your health information with the research team. Clinical trials need to function according to federal privacy laws, mainly the Health Insurance Portability and Accountability Act (HIPAA). You will have the opportunity to authorize any sharing of your personal information and understand how your information might be used during the informed consent process.
Typically, medical information for the purposes of a clinical trial (for example, laboratory test results, information about side effects) is entered into a database and shared only with the people who analyze the study results. Before any of your medical information is entered into the study database, your name is removed and your records are given a code or number. In some cases, members of the research team or regulatory authorities (for example, the FDA) may need to review these records; however, your personal information will not be shared with them. If the researchers publish the results of the clinical trial, they will never use your personal information.
What happens if I decide to leave the trial?
You can leave the trial—withdraw from the study—at any time. Let your research team, including your doctor, know if you have any questions or concerns. Talk to your doctor so you can discuss how leaving the trial might affect your health. Deciding to leave a trial will not affect your regular medical care.